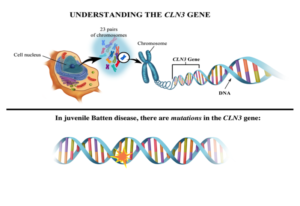
 The juvenile Batten disease gene and its protein
The juvenile Batten disease gene and its protein
The CLN3 gene and protein “What we know”
One of the most important steps taken by Beyond Batten Disease Foundation in our quest to cure juvenile Batten (CLN3) disease is to document the experimental history of the CLN3 gene and its corresponding protein. We believe that a strong understanding of where we are in terms of their regulation, structure, distribution lays the groundwork for determining therapeutic action plans.
BBDF compiled all of the available CLN3 gene and protein data from biological databases, repositories of federally and privately funded projects, patent and trademark offices, science and technology journals, industrial drug and pipeline reports as well as clinical trial reports and, with meticulous precision, validated the information together with experts in Batten disease from laboratories across the globe. To find a copy, click here.1 Here are a few highlights:
The juvenile Batten disease gene
Juvenile Batten disease results from mutations (mistakes) in the CLN3 gene (blueprint) responsible for making CLN3 protein. More than 67 different mutations in the CLN3 gene have been shown to cause juvenile Batten disease.2 However, most children with the disease are missing the same string of 966 base pairs (DNA building blocks) in one or both of their mutated copies of the CLN3 gene; this mutation is known as the “1kb deletion.” The effects of the 1kb deletion and other disease-causing mutations are not well-understood, although it is believed that such a large missing piece of the blueprint for CLN3 means cells aren’t making fully functional CLN3 protein.1
CLN3 Protein
The full-length CLN3 protein is predicted to be a 438 amino acid, integral, 6-pass, transmembrane protein. In other words, it resides in the membranes or walls of various cellular organelles or compartments looping in and out of the membrane-like sewing stitches through fabric.
The sequence does not display significant similarities to any protein of known function yet appears to contain a mitochondrial (cell battery) targeting site and two lysosomal targeting motifs.1 The identical full-length protein can be synthesized from two different mRNAs transcribed by specific and alternatively regulated promoters GenBank, UniProtKB/Swiss-Prot, GenBank.3
Where is CLN3 protein supposed to be?
CLN3 protein is found in the membranes (walls) of various organelles (compartments). Much like a bustling city with departments for utilities and transportation, organelles have different functions while working together to support the cell’s overall health. There are mitochondria (power plants), Golgi apparatus (processing and packaging centers), endoplasmic reticulum (transit centers), and garbage disposal and recycling centers (lysosomes). In neurons, CLN3 protein can be found in special compartments called synaptosomes, which are essential for communication between nerve cells. CLN3 may have the same function in every compartment of a cell, different functions in each, or a mixture.1
What is CLN3 protein doing in all of these places?
The maturation of cellular vesicles, cellular transport, and cellular cargo distribution are all disrupted in CLN3-deficient cells leading to an accumulation of undigested waste material, the pathologic hallmark of CLN3 disease, and evidence for lysosomal function(s) of CLN3 protein. Decreased levels of 28 lysosomal soluble and 11 lysosomal lipid-degrading enzymes in patient cells suggests that CLN3’s lysosomal function could be to transport various degradative enzymes into the lysosome where waste is destroyed and recycled. If so, this could explain why CLN3 protein is found in Golgi apparatus processing and packaging centers, endoplasmic reticulum transit centers, and sanitation department lysosomes. It could also explain neuron-specific cell death as transport and accumulation defects affect the release of neurotransmitters (specialized chemicals in the brain) important for communication between and the survival of neurons.3
The presence of CLN3 protein in mitochondrial membranes (cellular batteries) along with reported mitochondrial deficiencies in patient cells and CLN3 disease models strongly suggests a role for CLN3 in the maintenance of high energy fuel levels. The absence of CLN3 could be partially or wholly responsible for the toxic levels of reactive oxygen species (ROS) one sees in patient cells leading to further damage of mitochondria.1,3 To learn about the State of therapeutic development in CLN3 disease, click here.
How do we figure out what CLN3 protein does? More importantly, how do we fix the problems that occur when functional CLN3 protein is missing?
University-based researchers are hard at work using a variety of state-of-the-art techniques and animal models to 1) pinpoint the function(s) of the CLN3 protein, 2) prioritize which CLN3 protein functions are most critical to the cell’s overall health and proper function, and 3) pair their findings and expertise with pharmaceutical scientists to develop therapeutic approaches to prevent the earliest and most damaging effects of the loss of CLN3 protein – a process estimated to take an average of $1.3 billion and 12 years to reach New Drug Approval to treat disease.4 To see an animation of our strategic path toward a cure, click here. To learn about the State of therapeutic development in CLN3 disease, click here.
References
- Mirza M, Vainshtein A, DiRonza A, et al. The CLN3 gene and protein: What we know. Mol Genet Genomic Med. 2019. Dec:7(12).
- NCL Resource: A Gateway for Batten Disease [Internet]. London: University College London; Available from: http://www.ucl.ac.uk/ncl/batten.html.
- Shematorova EK and GV Shpakovski. Current Insights in Elucidation of Possible Molecular Mechanisms of the Juvenile Form of Batten Disease. Int J Mol Sci 2020 Nov; 21(21):8055.
- DiMasi JA, Grabowski HG, and RW Hansen. Innovation in the pharmaceutical industry: New estimates of R&D costs, J Health Econ. 2016. Vol 47, P20-33.